Introduction
Acute myeloid leukaemia (AML) is the most common form of acute leukaemia in adults. The average age at the time of diagnosis is 68 years. Median survival is highly dependent on the genetic risk profile, the age and fitness of the patient, and the possibility to administer potentially curative therapy, such as intensive chemotherapy or allogeneic stem cell transplantation. While the prognosis in younger patients has improved significantly over the past decades, the relative 5-year survival in older patients lags behind. This is partly because older patients with AML are not always fit enough for potentially curative intensive therapy, and partly because they are more likely to have an AML with genetic features associated with an unfavourable prognosis.
Thanks to significant developments in the diagnosis and treatment of AML, even for those patients who aren’t fit for intensive therapy, additional treatment options are becoming available and more effective combination therapies are leading to better outcomes. Some of these combination therapies include novel agents that specifically target mutations present, for example mutations in FLT3. These mutations may already be present at diagnosis, but may also arise later in the disease process.
It is therefore important to monitor the presence of mutations or their emergence during the disease or upon relapse. Increasingly, measurable residual disease (MRD) is being used for this purpose. With these developments, the treatment of AML is becoming more individualised and better tailored to the patient.
This Pharmacotherapy Online publication discusses the most important developments in the diagnosis and treatment of AML, using the latest national and international AML guidelines. New treatment options are illustrated using relevant case histories.
Clinical picture
AML is characterised by genetic changes in the DNA of stem cells and haematopoietic progenitor cells in the bone marrow. The importance of some common genetic mutations is further discussed in the 'Pathophysiology' and 'Diagnostics' sections.
Changes in DNA cause a maturation and differentiation disorder in immature haematopoietic cells, leading to an increase in clonal and undifferentiated cells, also called blasts. These cells no longer respond to inhibitory signals from the bone marrow microenvironment and continue to proliferate uncontrollably without differentiating into more mature myeloid cells. This causes disruption of normal haematopoiesis, leading to anaemia, thrombocytopenia and neutropenia; patients may experience symptoms such as fatigue, recurrent or persisting infections, and bleeding. Other, more atypical symptoms include pain in bones and joints, enlarged lymph nodes, a full feeling in the abdomen and unexplained weight loss or loss of appetite.1
On initial disease presentation, due to a strong leukocytosis, symptoms may arise due to hyperviscosity, such as vision impairment, headache and shortness of breath. This phenomenon is called leukostasis and is an indication for immediate intervention with emergency chemotherapy.2
Epidemiology
Around 800 patients are diagnosed with AML annually in the Netherlands, slightly more often men (58%) than women (42%). Almost half of patients are 70 or older at diagnosis. Due to an ageing population, the incidence of AML has been increasing in recent years, from about 400 cases per year in the early 1990s to about 800 cases per year today.3
The relative 5-year survival of patients with AML up to age 70 rose significantly between 1989 and 2018 (see Figure 1). As standard intensive chemotherapy treatment has remained virtually unchanged since the 1970s, this improvement is mainly due to better supportive treatments (antibiotics, antifungal drugs, erythrocyte/thrombocyte transfusions, intensive care medicine, paramedical care) and improved care around stem cell transplants.
There is a clear relationship between a patient's age and their relative 5-year survival, with older patients generally having worse survival. In patients with AML over age 70, the low relative 5-year survival remains unchanged from 30 years ago (see Figure 1). This is partly because older patients are generally not (or not always found to be) fit enough for potentially curative intensive therapy (including stem cell transplantation) and partly because older patients are more likely to have AML with genetic features that predict poor risk (see also the 'Diagnostics' section).3

Figure 1 Relative 5-year survival of AML in different age groups.
Source: IKNL3
Pathophysiology
Over the past decades, many genetic abnormalities have been found that play or could play a role in the development of AML. These changes in DNA disrupt various processes within haematopoietic stem cells, such as cell division, differentiation, self-renewal and programmed cell death. Sometimes a single mutation or change is enough to lead to disease, but usually several changes are needed that combine to cause leukaemia.
Large research projects using whole-exome sequencing in AML patients have led to the discovery of many mutations, some of which may have important clinical implications, for example for the prognosis or treatment of patients. The eight most relevant mutations are discussed in more detail in the rest of this section.4
Specific inhibitors have been developed against a number of mutations, for example against mutated FLT3, IDH1 and IDH2. If the diagnostic examination shows that a patient with AML has such a mutation, a specific inhibitor can be used for it in certain cases. This is sometimes done in study settings, but also increasingly outside study settings (see also the 'Treatment' section).1
FLT3
Genetic alterations in FLT3 (mFLT3) occur in about 30% of AML cases. The gene FLT3 is located on chromosome 13 and is a type III receptor tyrosine kinase with important functions in haematopoietic cell survival, cell division and differentiation. In the context of AML, FLT3 has two different types of mutations:4
- FLT3-ITD: internal tandem duplication in the juxtamembrane domain. Part of the DNA has incorrectly multiplied, producing a constantly active and signalling protein. This mutation occurs in 25% of AML patients.5
- FLT3-TKD: point mutation or deletion in the tyrosine kinase domain. Part of the DNA has been incorrectly deleted, producing a constantly active and signalling protein. This mutation occurs in 7-10% of AML patients.5
Historically, only the FLT3-ITD mutation was associated with an unfavourable prognosis and a high risk of relapsed AML.5 Since the advent of pharmacological inhibitors of FLT3 such as midostaurin, gilteritinib and quizartinib, the prognosis of patients with mFLT3-AML has slightly improved, so this mutation is now classified as intermediate-risk (see also the 'Treatment' section).6
IDH1/IDH2
In about 20% of patients with AML, mutations occur in IDH1 or IDH2. IDH1 and IDH2 are enzymes involved in metabolic and epigenetic cellular processes. Mutations in these enzymes (mIDH1/2) cause DNA hypermethylation, aberrant gene expression and a differentiation blockade. Various publications present the effect of mIDH1/2 on the prognosis of AML patients as ambiguous and also dependent on additional genetic abnormalities.4 A selective inhibitor of IDH1, ivosidenib, has recently been approved and is now reimbursed in primary care for patients with mIDH1 (see also the 'Treatment' section). Olutasidenib, which is also a mIDH1 inhibitor, has now been approved in the US but is not yet available in Europe. The same applies to enasidenib, a selective inhibitor of mIDH2.
NPM1
NPM1 is mutated in about 30% of patients with AML.4 NPM1 is located on chromosome 5 and is a multifunctional phosphoprotein with various regulatory functions within the cell. The tumour-suppressor genes CDKN2A, TP53 and MDM2 are among those under the control of NPM1.4
Unlike the above-mentioned mFLT3, mutated NPM1 (mNPM1) is associated with a relatively favourable prognosis, but only in the absence of a co-mutation in FLT3 and the absence of poor prognostic cytogenetic abnormalities. Studies have shown that patients with mNPM1 are more likely to achieve complete remission, have a better general survival and are less likely to develop relapsed AML than patients with wild-type NPM1.7
CEBPA
A mutation in CEBPA occurs in about 10-20% of AML cases. CEBPA is located on chromosome 19 and is a transcription factor that regulates myeloid differentiation. Genetic alterations can occur in one allele (moCEBPA) or both alleles (biCEBPA) of the gene. Both variants are about equally common.8
biCEBPA mutations are associated with a relatively favourable prognosis, and in 90% of cases this involves biallelic mutations in the leucine zipper region of the gene (bZIP). For moCEBPA mutations this is less clear, as there are several variants. A mono-allelic bZIP mutation occurs in only 35% of patients with a moCEBPA mutation, but in this case it is associated with a relatively good prognosis. For patients with a mono-allelic mutation in the N-terminal transactivation domain (TAD), the prognosis is less favourable. BiCEBPA and bZIP mutations share a number of characteristics. These mutations are particularly common in younger patients, and certain co-mutations are common in both cases, such as mutations in GATA2 and NPM1.9
CEBPA mutations have also been implicated in familial forms of AML, which often occur early in life and are associated with a relatively favourable prognosis.10
RUNX1
RUNX1 is located on chromosome 21 and is a transcription factor, responsible for regulating critical processes in the differentiation during haematopoiesis. RUNX1 can mutate (mRUNX1) but can also be involved in a chromosomal translocation, both in AML and in other forms of leukaemia.4
One of the most common translocations in AML is t(8;21)(q22;q22.1). This creates a fusion protein between RUNX1 and ETORUNX1T1. This fusion protein is found in about 12% of all AML patients and is associated with a relatively favourable prognosis.11 In contrast to this chromosomal translocation involving RUNX1, mRUNX1 often has an unfavourable outcome. mRUNX1 can also occur familially and, in addition to an increased risk of developing AML/MDS, is also associated with thrombocytopenia and thrombocytopathy.
ASXL1
Mutations in ASXL1 (mASXL1) occur in 15-20% of all AML patients. Mutations in this gene are up to five times more common in older patients (aged ≥60) than in younger patients (aged <60).12 ASXL1 is located on chromosome 20q11 and is an epigenetic modulator. mASXL1 disrupts haematopoiesis and stimulates myeloid transformation. mASXL1 often accompanies trisomy 8, abnormalities on chromosome 11, and mRUNX1 or mIDH2. A mutation in ASXL1 is associated with an unfavourable prognosis.13
TP53
TP53 mutations (mTP53) are present in <10% of patients with new AML, but are more common in patients with relapsed or refractory AML because they are associated with resistance to various treatments and therefore associated with a very poor prognosis. TP53 is located on chromosome 17p and is a transcription factor with important functions in repairing DNA mismatches and repairing base and/or nucleotide deletions. AML is in rare cases caused by a familial heterozygous mTP53 n the context of Li-Fraumeni syndrome.14
KMT2A
Mutations in KMT2A (mKMT2A) occur in about 3% to 11% of AML patients, particularly in people with normal cytogenetics or trisomy 11 as the only cytogenetic abnormality, and in that case involve a partial tandem duplication in KMT2A.15 These mutations have no significant value for the prognosis of AML, except if KMT2A, which lies on chromosome 11q, is involved in a chromosomal translocation. The most common translocation is t(9;11)(p21.3;q23.3), in which a fusion protein occurs between KMT2A and MLLT3. This translocation is associated with an intermediate prognosis. Other translocations involving KMT2A (over 100 different forms have been described) are associated with an unfavourable prognosis.16,17
Through clonal haematopoiesis, genetic mutations accumulate throughout life. Some mutations, such as mASXL1 and mIDH1/2, usually arise early in the development of AML and are therefore found in almost all clonal cells. Other mutations, such as mNPM1 or mFLT3, occur secondarily and are therefore present in a smaller portion of clonal cells, unless the subclone in which the mNPM1 or mFLT3 developed has displaced or inhibited the other clones due to a competitive growth advantage.18
Diagnostics
Diagnostics in AML serve several purposes. First and foremost, diagnostics when acute leukaemia is suspected should confirm the diagnosis. In addition, diagnostics are necessary to determine the subtype of leukaemia and identify the patient's risk profile, in order to decide on the best treatment.
Confirm diagnosis
To confirm the diagnosis of AML, a case history combined with physical examination and a complete blood count and bone marrow testing are indispensable.1
The case history should pay special attention to familial cancer predisposition syndromes and haematological abnormalities. During the physical examination, attention should be paid to:1
- Skin abnormalities
- Lymphadenopathy
- Splenomegaly
- Gingival hypertrophy
- Testis enlargement
- Fever
- Mouth/teeth
Infection sites (especially perianal region)
- Headache
- Neurological deficit or other disorders
- Abnormalities associated with hereditary predisposition:
- Warts
- Lymphedema
- Early greyness
- Abnormalities of the oral mucosa or nails
- Hypo-/hyperpigmentation of the skin
- Leukoplakia
The blood and bone marrow tests serve to morphologically determine the number of blasts. In addition, immunophenotyping can be used to determine whether the leukaemia is of myeloid origin (AML), lymphoid origin (ALL), undifferentiated origin (acute undifferentiated leukaemia, AUL) or mixed origin (mixed phenotype acute leukaemia, MPAL). Last, genetic diagnostics can be performed on blood and/or bone marrow, which can reveal cytogenetic abnormalities or mutations through molecular diagnostics.
Classification criteria for AML
AML can be diagnosed based on criteria established by the WHO and the International Consensus Classification (ICC). There are minor differences between these classifications, but in essence they are very similar.19 AML can be diagnosed if at least one of the following characteristics is met:1
- ≥20% myeloid blasts in blood or bone marrow
- Presence of AML-defining genetic abnormalities, including t(8;21), t(15;17), inv(16), t(6;9), inv(3), t(9;11), mNPM1, mCEBPa (for WHO, regardless of percentage of blasts, for ICC ≥10% blasts required)
The diagnostic criteria of both the ICC and WHO have a number of subgroups, including AML with mutated TP53, AML with myelodysplasia-related gene mutations and AML with chromosomal abnormalities. In addition, in both classifications there is an intermediate category of patients with myelodysplastic syndrome (MDS/AML in ICC and MDS-IB2 in WHO).19 These are patients with 10-19% blasts and clinical outcomes similar to patients with a higher number of blasts who actually do have AML. Several studies have shown that MDS with mNPM1 can develop into AML in a short time. This is why this group is now included in the diagnostic criteria for AML.
Risk rating
The European LeukemiaNet (ELN) risk classification is used to get a picture of prognosis and thus determine treatment.
As mentioned in the 'Pathophysiology' section, different genetic disorders are associated with different prognoses. The prognoses of various cytogenetic abnormalities are defined in the ELN risk classification. There are three risk groups: (relatively) favourable, intermediate and adverse. Which cytogenetic abnormalities belong in which risk group is shown in Table 1. These risk profiles are based on studies with intensively treated patients and therefore do not apply well to patients treated with non-intensive therapy such as hypomethylating agents, whether in combination with venetoclax or not.
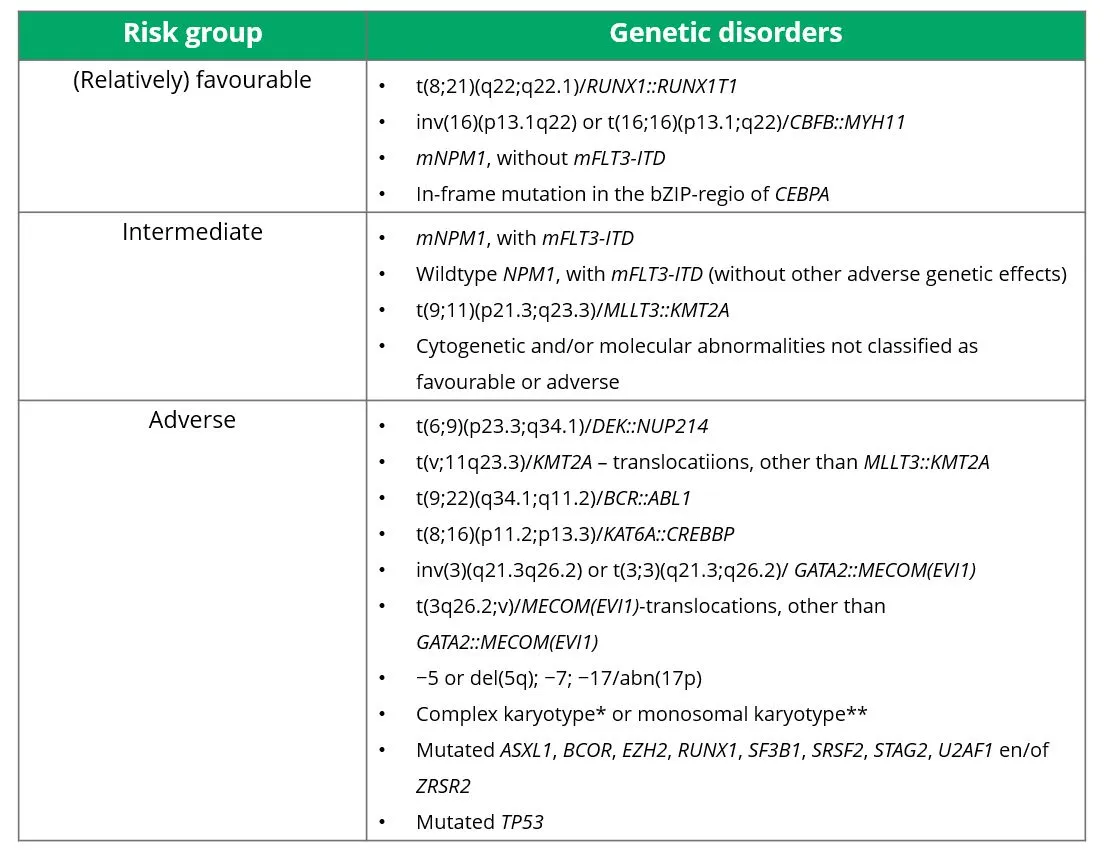
Table 1 Risk groups for AML, according to the 2022 ELN criteria.
* Complex karyotype = ≥3 unrelated chromosomal abnormalities, in the absence of any of the genetic disorders from the WHO category 'defined genetic disorders'.
** Monosomal kayotype = ≥2 different monosomies (excluding X or Y) or 1 autosomal monosomy combined with at least 1 structural chromosomal abnormality (excluding core-binding factor AML)
Source: Döhner, et al. 2022.20
Response criteria for AML
During treatment of AML, blood or bone marrow tests should be repeated to check whether the disease has achieved remission. This is usually done at a minimum after each cycle of intensive chemotherapy. In non-intensive therapy, the speed to response is highly dependent on the regimen chosen, and thus the risk of bone marrow toxicity should be weighed at the time of bone marrow evaluation. For example, when treating with hypomethylating therapy, without additional agents, the duration to response will be longer on average and there is less risk of bone marrow toxicity. A bone marrow test can then be done after three courses. However, adding venetoclax to the treatment will shorten the duration to response to 1-2 cycles on average, but also increases the risk of bone marrow toxicity. Therefore, in this regimen bone marrow testing is recommended as early as 21 days after starting. Different criteria exist to reflect response to treatment. These response criteria are summarised in Table 2.20

Table 2 Response criteria for assessing AML after treatment.
* In patients with CR, CRh or CRi, a low percentage of circulating blasts may be present in the blood. This may be a sign of bone marrow recovery. If this is the case, the circulating blasts disappear within a week and there is no persistent disease.
Source: Döhner, et al. 2022.20
MRD
Measurable Residual Disease (MRD) is an important biomarker in AML and is used for prognostic purposes, monitoring and efficacy-response measurements. Advanced flow cytometric and/or molecular techniques can detect residual AML cells with greater sensitivity, and thus have a better level of detection than cytomorphology. The cornerstone of achieving complete remission (CR) is having <5% blasts in the bone marrow. This morphological technique thus has an arithmetic sensitivity of 5x10-2 cells. However, as there are different techniques to measure MRD, a depth of 1x10-7 can be achieved, depending on the technique.
Although MRD is clinically relevant and already widely used in daily practice, its determination and interpretation remain complex. Several methods are available, briefly described below.21
MFC-MRD test
Measuring MRD using multiparameter flow cytometry (MFC) is the most commonly used method. In MFC-MRD, proteins on the surface of bone marrow cells or blood cells are detected and visualised using antibodies to which a fluorescent marker is attached. MRD identification takes advantage of the fact that AML cells often show immunophenotypic abnormalities. What’s important here is that the abnormalities are different from those of normal cells. For example, antigens on leukaemia cells can:
- Be absent, while expressed on their normal counterpart (e.g. absence of CD33)
- Be expressed, while never present on normal cells (e.g. lymphoid antigens on myeloid cells)
- Not match the maturation stage of the cells (e.g. antigens of mature cells present on progenitors)
Together, these aberrant antigens form the leukaemia-associated immunophenotype (LAIP). AML is highly heterogeneous, and the kind and type of LAIPs vary from case to case. Therefore, it is necessary to deploy a comprehensive and standardised immunophenotyping panel. This allows LAIP to be identified in about 90% of patients.21
Molecular MRD test
A molecular MRD test can be performed via polymerase chain reaction (PCR) or next-generation sequencing (NGS). To achieve sufficient limit-of-detection, it is necessary to use validated techniques such as real-time quantitative PCR (qPCR), digital PCR (dPCR) or error-corrected NGS.
PCR is particularly useful when one or more mutations or transcripts are present whose DNA sequence is fully known. This is the case in about 40-60% of patients with AML. These include mutations in: NPM1, RUNX1::RUNX1T1, CBFB::MYH11, PML::RARA, KMT2A::MLLT3, DEK::NUP214, BCR::ABL and WT1. For PCR analysis, leukaemia-specific mutations such as NPM1 PML::RARA, RUNX1::RUNX1T1 and CBFB::MYH11 are preferred to less specific mutations.21
Both peripheral blood and bone marrow can be used for MRD, but the specificity of PCR is 5-10 times lower in peripheral blood than in bone marrow. The choice of blood or bone marrow depends on the time and purpose of evaluation. When WT1 is the only mutation that can be investigated, it is recommended to perform this on peripheral blood, as bone marrow cells naturally have higher WT1 expression than cells in peripheral blood and the test may therefore be falsely positive.21
In patients with mutations whose DNA sequence is not fully known, MRD can be determined using NGS. The advantage of this technique is that mutations known from diagnosis can be tracked. It can also detect newly developed mutations. A drawback is that standard NGS has limited sensitivity (tenfold to hundredfold lower than PCR) and is therefore not yet accepted as a valid MRD method in the current MRD consensus guideline. However, sensitivity will improve rapidly in the future with the advent of error-corrected NGS.
Almost all detected mutations could theoretically serve as MRD markers,but this does come with some limitations. Germline mutations (which in the context of AML can occur in, for example, the genes ANKRD26, CEBPA, DDX41, ETV6, GATA2, RUNX1 and TP53) are not suitable as a marker, as these mutations are also present in non-malignant cells and therefore do not provide information on MRD unless an allogeneic stem cell transplant has been performed. In addition, mutations may be associated with age-related non-malignant clonal haematopoiesis. Best known examples are mutations in DNMT3A, TET2 and ASXL1 (DTA mutations) and thus do not provide information on the nature and quantity of leukaemic cells. In addition to DTA mutations, there is evidence that mutations in IDH1, IDH2 and SRSF2 may fit residual non-malignant clonal haematopoiesis. Outside the context of follow-up after allogeneic transplantation, these mutations should be disregarded in MRD analysis. If only mutations in these genes are present, the recommendation is to use MFC to determine MRD.21
According to the current guidelines, mutations in genes involved in signalling pathways (such as mutations in FLT3-ITD, FLT3-TKD, KIT, KRAS and NRAS) have little predictive value and thus should be combined with other markers to determine MRD.21 However, studies have shown that FLT3-ITD can be a valuable prognostic marker for AML. In 161 AML patients with an FLT3-ITD mutation, MRD status was determined both at diagnosis and in complete remission after intensive induction therapy. FLT3-ITD MRD was present in 47 patients (29%). The presence of FLT3-ITD MRD is associated with an increased risk of relapsed AML (incidence of 4-year cumulative recurrence was 75% in FLT3-ITD MRD positivity and 33% in FLT3-ITD MRD negativity, p<0.001) and inferior overall survival (4-year overall survival was 31% in FLT3-ITD MRD positivity and 57% in FLT3-ITD MRD negativity, p<0.001).22 Detection of FLT3-ITD MRD at the time of complete remission may be prognostically important, especially since drugs specifically acting against the FLT3-ITD mutation have recently become available (see also the 'Treatment' section and Case III).
MRD test results
An MRD test can give a positive or negative result. Definitions of a positive MDR test are given below by method:21
- MFC-MRD is positive if ≥0.1% of CD45+ cells express the sought-after immunophenotype
- PCR-MRD is positive at a cycle threshold <40 in ≥2 or 3 replications
- NGS-MRD is positive with variant allele frequency ≥0.1%
A negative test result doesn’t mean that no residual disease is present, but that the value falls below the chosen clinical or technical cut-off value of the test. Below this value, residual disease may well be present. When residual disease is detected but falls below the clinical cut-off value, molecular testing speaks of MRD at low level of MRD-LL. For MRD-LL, the value found is <2% lower (for NGS-MRD) or higher (for PCR-MRD) than the threshold. So there is no positive result, but the value is well above the detection limit of the test. The exact prognostic value of MRD-LL is not yet fully understood.21
MRD response or relapse
Based on the results of the MRD test, a classification can be made into different response categories. These are shown in Table 3.

Table 3 Response categories in determining MRD.
Source: Heuser, et al. 2021.21
Clinical implications of MRD testing
Failure to achieve MRD-negative remission in MFC-MRD, molecular MRD positivity after consolidation therapy and MRD recurrence are associated with a higher risk of recurrent AML and inferior outcomes. The following groups of patients are eligible for individualised treatment and/or combination therapies (options are discussed in more detail in the 'Treatment' section):21
- Patients with positive MFC-MRD after two cycles of intensive chemotherapy, after consolidation therapy, and before or after allogeneic stem cell transplantation
- Patients with an mNPM1 who remain MRD-positive after consolidation therapy with ≥2% mNPM1 copies per ABL1 copy or in whom no 3/4-log decrease is apparent in mNPM1 between diagnosis and termination of consolidation therapy (measured in the same tissue, preferably bone marrow)
- Patients with MRD relapse
Figure 2 shows for each AML phenotype how and when MRD studies are best used.

Figure 2 Recommendations for the deployment of MRD testing in different AML phenotypes.
Based on: Heuser, et al. 2021.21
Treatment
The goal of AML treatment is to control and, if possible, cure the disease. Ideally, complete remission is achieved on initial therapy (induction therapy). With consolidation or maintenance therapy, that remission can then be further deepened to reduce the risk of relapse. Crucial to starting the right therapy are the results of genetic analyses. These should preferably be available within three to five days of diagnosis. If genetic analysis results take longer to come back, it is wise to delay therapy. A short delay in starting treatment in clinically stable patients has no adverse effect on final clinical outcomes.20,23
In patients who cannot tolerate potentially curative therapy, the treatment goal should be to optimise quality of life, prevent complications resulting in transfusion or other supportive care measures and involve palliative care providers in a timely manner.20
Treatment of patients fit for intensive therapy
Treatment of patients fit for intensive therapy is discussed below and summarised schematically in Figure 6.
Induction therapy
Cytarabine and anthracyclines (such as daunorubicin and idarubicin) form the backbone of intensive chemotherapy. In most schedules, cytarabine is administered for seven days and anthracyclines for three days, which is why this schedule is also called ‘7+3’. For patients with mFLT3, the kinase inhibitor midostaurin is now part of standard treatment, as an adjunct to induction treatment.
Cytarabine
Cytarabine, also known as cytosine arabinoside or Ara-C, is a chemotherapeutic drug belonging to the class of antimetabolites. The mechanism of action of cytarabine involves its incorporation into DNA during the S phase of the cell cycle, where it acts as a synthetic analogue of the nucleoside cytidine. Once incorporated into the DNA molecule, cytarabine disrupts DNA strand synthesis, which can prevent further DNA replication and repair. This disruption eventually leads to cell death, especially in rapidly dividing cells such as AML cells.
Because of the pharmacodynamic principle that rapidly dividing cells are more selectively killed by continuous low exposure to chemotherapy, cytarabine is given as a continuous infusion in the first remission induction course. As a result, these rapidly dividing cells are all exposed to cytarabine at least once in their S phase within the seven-day administration period. In the second remission induction treatment, cytarabine is administered as a high dose, or ‘pulse’, twice daily, which could also kill slowly dividing AML cells. This variation in cytarabine administration may be particularly relevant for patients who have shown a suboptimal response after the first induction course, but it is not based on clinical evidence with hard endpoints.24
Anthracyclines
Anthracyclines are chemotherapeutics used in multiple tumour types and are topoisomerase II inhibitors. In the case of AML, daunorubicin is mostly used in the Netherlands, while idarubicin is also widely used abroad. The mechanism of action of anthracyclines relies on several aspects. First, they interfere with DNA metabolism by interacting with nucleic acids, resulting in inhibition of DNA and RNA synthesis and suppression of cell proliferation, especially in rapidly dividing cells such as cancer cells. Second, anthracyclines cause oxidative damage by forming reactive oxygen species, such as free radicals, which in turn cause damage to cell membranes and DNA. This dual action contributes to the cytotoxic effects of anthracyclines on cancer cells.
Due to cardiotoxicity, there is a maximum cumulative anthracycline dose that can be given. For this reason, anthracyclines are rarely used in relapsed AML, as patients in the first line of treatment will often have been extensively exposed to anthracyclines already. Also, in patients who have previously been treated with anthracyclines for another type of cancer and are subsequently diagnosed with AML, for example in the context of therapy-related AML, an anthracycline-sparing regimen may need to be diverted to an anthracycline-sparing regimen in order not to cause a significantly increased risk of heart failure.
Midostaurin
The efficacy of midostaurin for the treatment of patients with mFLT3 was investigated in the RATIFY study (phase III). Patients aged 18-59 with mFLT3 AML could take part in the study. They were randomised across two groups: a midostaurin group (n=360) and a placebo group (n=357). Both groups were treated with standard induction and consolidation therapy. To this either midostaurin or a placebo was added. The primary endpoint of the study was overall survival. The secondary endpoint was disease-free survival.25
Both overall survival and disease-free survival were significantly higher in the midostaurin group than in the placebo group (overall survival: hazard ratio 0.78, p=0.009; see also Figure 3a; disease-free survival: hazard ratio 0.78, p=0.002). The number of serious adverse events was similar in both groups. The beneficial effect of midostaurin was evident in all patients with mFLT3: mFLT3-ITD high (n=214), mFLT3-ITD low (n=341) and mFLT3-TKD (n=162). The subgroup analysis is shown in Figure 3b.25

Figure 3a (top) Mean overall survival of midostaurin (green) and placebo (red), from randomisation to 90-month follow-up.
Figure 3b (bottom) Subgroup analysis of patients with mFLT3 AML. In all groups, treatment with midostaurin was more favourable than placebo.
Source: Stone, et al. 2017.25
Gilteritinib
In the context of a clinical trial (HOVON 156), until 2023 it was also possible to give the FLT3 inhibitor gilteritinib, in combination with intensive chemotherapy. The study included the maximum number of patients in 2023, and results are now awaited.
Gemtuzumab ozogamicin
In addition, there is the possibility of adding gemtuzumab and ozogamicin (GO) to treatment for patients with newly diagnosed CD33+-AML. This addition improves survival in patients with core-binding factor AML (t(8;21), inv(16) or t(16;16)) or an NPM1 mutation in several studies. Because these studies were not performed using the standard HOVON induction schedule, it is not known whether adding GO in this setting also adds value. GO is therefore not a widely used tool in the Netherlands.1
CPX-351
An alternative to standard induction therapy is treatment with Vyxeos (CPX-351). Vyxeos is a liposomal form of daunorubicin and cytarabine in solid 1:5 molar ratio. It is registered for the treatment of therapy-related AML and AML with myelodysplastic changes. The Dutch guideline recently concluded that there is no good data showing that Vyxeos has added value over the standard HOVON induction regimen. The use of Vyxeos is therefore not recommended.1
The options and doses for induction therapy in fit patients are shown in Table 4.1,20
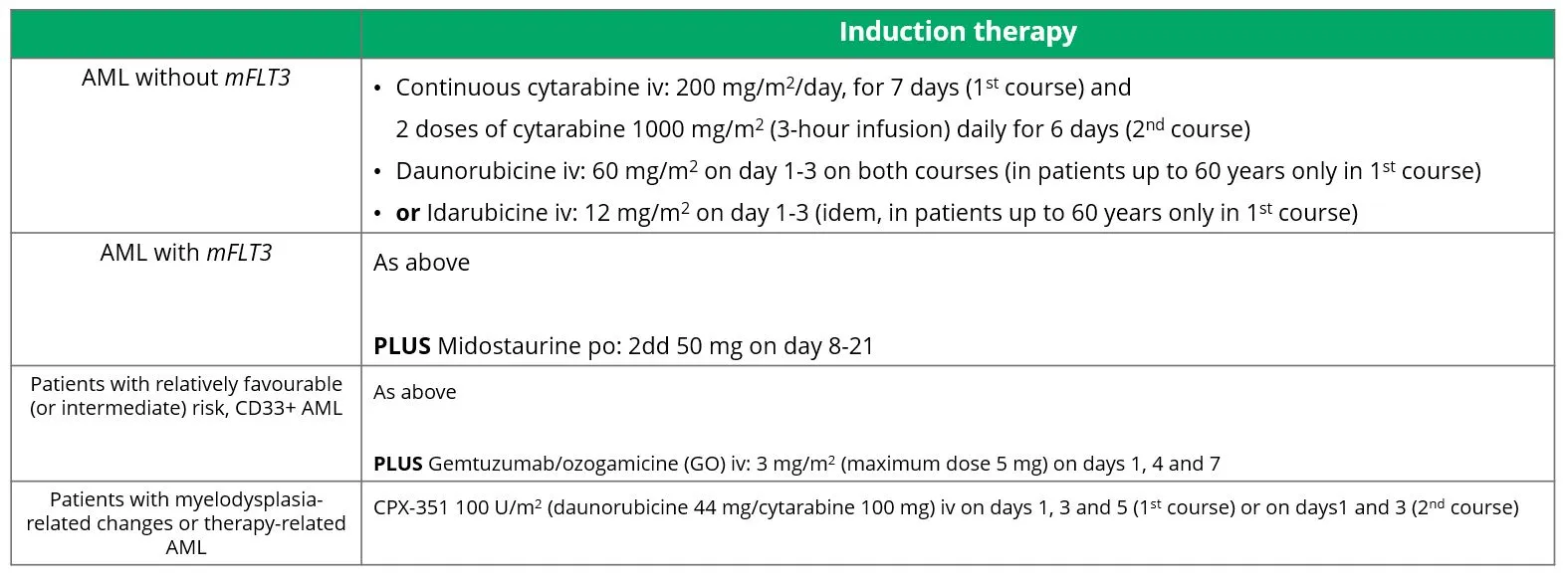
Table 4 Induction therapy in the treatment of AML.
Source: Döhner, et al. 2022.20
In principle, patients receive a second remission induction course after the first remission induction course and subsequent repopulation, unless unacceptable toxicity has occurred during or after the first remission induction course.
Consolidation therapy
After reaching CR, CRh or CRi, a switch can be made to consolidation therapy. If no CR, CRh or CRi is achieved after two cycles of intensive induction therapy, it is possible to switch to second-line treatment. Options for this are discussed further under the heading 'Treatment for refractory disease or relapsed AML'.
The consolidation treatment depends on the risk profile and the presence of MRD (see ELN risk classification, Table 1).
Within HOVON, patients with a favourable cytogenetic risk profile are consolidated with myeloablative chemotherapy (busulfan and cyclophosphamide) and autologous stem cell reinfusion after achieving complete remission, regardless of MRD status. In some patients, however, it is not possible to harvest stem cells. In these cases, a third course of mitoxantrone etoposide is an option. However, sometimes the choice is also made to consolidate with 3-4 courses of intermediate-dose cytarabine or allogeneic stem cell transplantation.1
In patients with an NPM1 mutation who have become MRD-negative or have achieved good reduction of mNPM1, autologous consolidation is also advised in principle. If there is insufficient reduction of mNPM1 or if there is an intermediate risk profile with presence of MRD, then the advice is to consolidate with allogeneic stem cell transplantation.1
In patients with an adverse risk profile, allogeneic transplantation is always recommended, regardless of MRD status. Higher age (>60-65 years) or failure to achieve early CR (remission after first course) are generally associated with a poorer prognosis and therefore factors that can be used to intensify consolidation (allogeneic rather than chemo-consolidation).1
Non-HOVON countries largely use other consolidation schemes. There are also special treatment regimens there for patients with a relatively favourable risk profile (especially CD-33 positive AML), AML with myelodysplasia-related changes or therapy-related AML, and patients with mFLT3.20
The different consolidation therapies are shown in Table 5.

Table 5 Consolidation therapy in the treatment of AML.
* At age >60-65 and/or failure to achieve early CR (CRe), allogeneic stem cell transplantation should be considered.
Source: HOVON, Guideline AML. 2021.1 Döhner, et al. 2022.20
Maintenance therapy
After completing induction and consolidation therapy, maintenance therapy follows in some cases. The aim of this treatment is to reduce the risk of recurrence. Midostaurin is frequently given to patients who have also been treated with this agent in induction and/or consolidation therapy, although no studies have yet been conducted on the use of midostaurin as maintenance therapy for AML.
Maintenance therapy with azacitidine subcutaneously or oral azacitidine can be considered for patients who nevertheless do not prove suitable for consolidation with intensive chemotherapy after intensive therapy or who are not (yet) eligible for allogeneic stem cell transplantation. Azacitidine subcutaneously can also be used to bridge to allogeneic stem cell transplantation.1,20
The various maintenance therapies with azacitidine are discussed further below.
Azacitidine subcutaneously
The efficacy of azacitidine subcutaneously was investigated in the HOVON97 study (phase III). Patients 60 and older with AML or myelodysplastic syndrome took part in the study. All patients were in remission (CR/CRi) after at least two cycles of intensive chemotherapy. Participants were randomised across two groups: azacitidine subcutaneously, 50 mg/m2 on days 1-5, every four weeks with a maximum of 12 cycles (n=56) or control group, observations only (n=60). The primary endpoint of the study was disease-free survival. The secondary endpoint was overall survival.26
After 12 months of follow-up, disease-free survival was significantly better in the azacitidine subcutaneous group than in the control group (64% versus 42%, respectively, log rank p=0.04; see also Figure 4). This difference persisted after adjustment for adverse-risk cytogenetic abnormalities and blood counts at randomisation. Mean overall survival did not differ between the two groups, probably because many patients in the control group underwent allogeneic stem cell transplantation after relapse. Rescue medication was used more often in the control group (32 times) than in the azacitidine subcutaneous group (9 times).26

Figure 4 Estimated disease-free survival of azacitidine subcutaneously (green) and control (red), from randomisation to 30-month follow-up.
Source: Huls, et al. 2019.26
Oral azacitidine
The efficacy and safety of oral azacitidine (CC-486) was investigated in the QUAZAR study (phase III). Patients aged 55 or older who were in first remission (CR/CRi) after intensive chemotherapy were randomised across two groups: CC-486 300 mg (n=238) or placebo (n=234), daily for 14 days per 28-day cycle. The primary endpoint of the study was overall survival.27
Mean overall survival was significantly higher in the CC-486 group than in the placebo group (24.7 months versus 14.8 months, respectively; p<0.001). Common adverse events in both groups were grade 1 and 2 gastrointestinal complaints. Grade 3 or 4 neutropenia was more common in the CC-486 group than in the placebo group (41% versus 24%, respectively).27
Even after more than two years of follow-up, the difference in mean overall survival remained visible between the two groups, albeit less pronounced than in the first period (Figure 5).28 The QUAZAR study has been criticised for suboptimal treatment of the control group, which might have increased the likelihood of study outcomes turning out in favour of CC-486.29
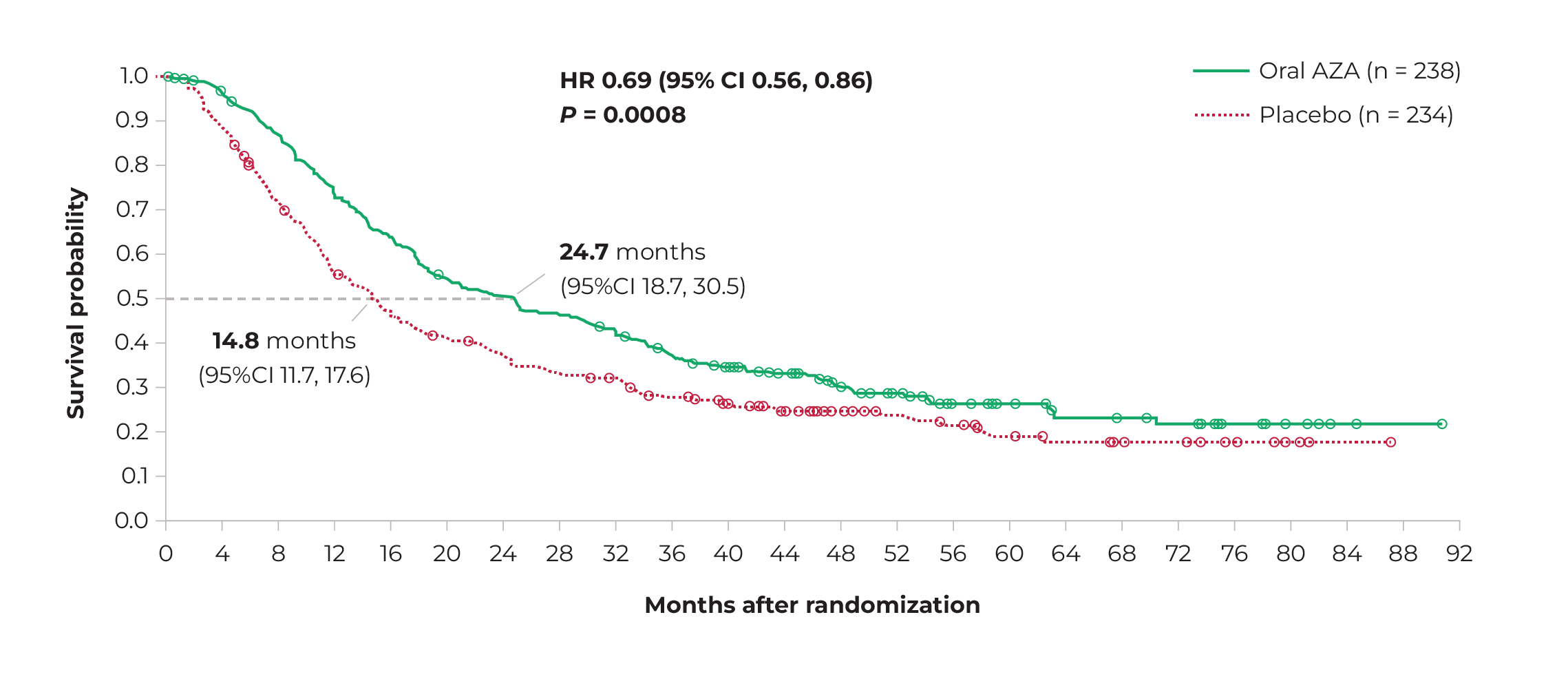
Figure 5 Estimated overall survival of oral azacitidine (green) and placebo (red), from randomisation to 92-month follow-up.
Source: Wei, et al. 2021.28
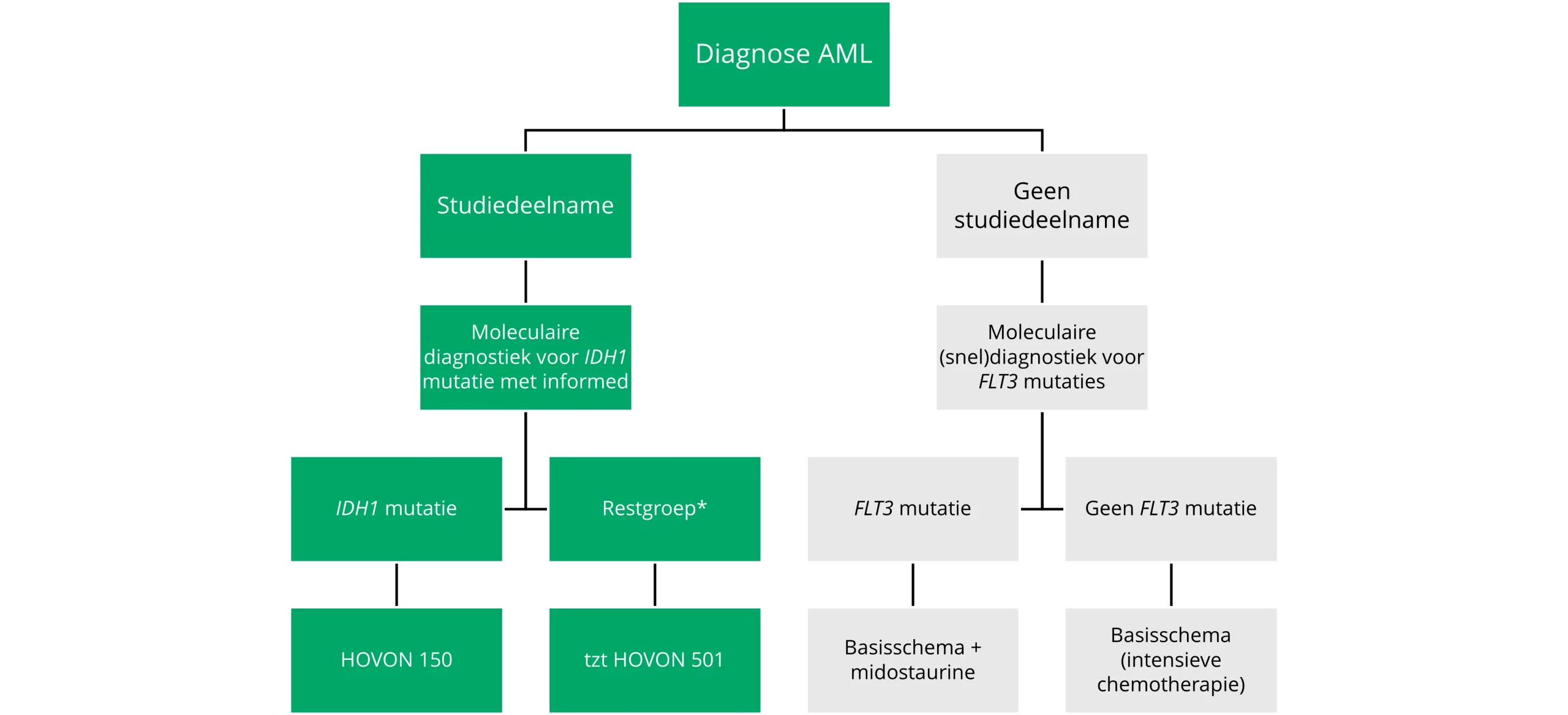
* With exception of FLT3 mutation.
Figure 6 Decision tree treatment for fit patients (varies by country).
Treating patients who are not fit for intensive therapy
To determine which patients are or aren’t fit for intensive chemotherapy, the Ferrara criteria (Table 6) are used internationally in research contexts.30 The Dutch guideline favours the Haematopoietic Cell Transplantation Co-morbidity Index (HCT-CI, see Table 7).1,31 This index includes various comorbidities with a score between 1 and 3. Patients over age 40 receive an extra 1 point above the total score due to their age.31 Patients with a score ≥3 (without including age) have about a 30% risk of death in the first 30 days of intensive treatment and are therefore found not fit more often. This does remain an individual trade-off, where patients with a high HCT-CI score are sometimes still being treated with intensive therapy, because with it a chance of curation can be aimed for. Still, the development of non-intensive treatment regimens has improved in recent years, providing better alternatives to intensive chemotherapy.

Table 6 Characteristics of patients not fit for intensive therapy according to the Ferrara criteria.
Source: Ferrara, et al. 2013.30

Table 7 HCT-CI score for assessing comorbidities for treatment of AML.
Source: Amsterdam UMC. Cellular therapy. Risk rating. HCT-CI.31
The standard treatment for patients who are not fit for intensive therapy is a combination therapy of azacitidine and venetoclax. For patients with mIDH1 AML, azacitidine and ivosidenib are given instead. Both treatment options are discussed in more detail below. Depending on patient characteristics and/or leukaemic features, it is also possible to opt for decitabine or azacitidine monotherapy. The next option (which is practically no longer given in the Netherlands) is cytarabine subcutaneously. As a final step, the patient is eligible for supportive treatment with e.g. hydroxycarbamide or 6-mercaptopurine, transfusions and/or antibiotics. All treatment options for non-fit patients are schematically summarised in Figure 9.1
Azacitidine + venetoclax
The efficacy and safety of azacitidine in combination with venetoclax were investigated in the VIALE-A study (phase III). Patients with AML who were ineligible for intensive chemotherapy due to age or comorbidities were randomised across two groups: azacitidine-venetoclax (aza-ven, n=286) or azacitidine-placebo (placebo group, n=145). In both groups, patients received a standard dose of azacitidine (75 mg/m2 SC/IV on days 1-7, on a 28-day cycle). Patients in the aza-ven group additionally received 400 mg venetoclax daily, orally; patients in the placebo group received similar placebo treatment. The primary endpoint of the study was overall survival.32
After a mean follow-up of 20.5 months, mean overall survival was 14.7 months in the aza-ven group and 9.6 months in the placebo group (hazard ratio 0.66; 95% CI, 0.52-0.85; p<0.001; see also Figure 6). The incidence of complete remission (CR) was significantly higher in the aza-ven group than in the placebo group (36.7% versus 17.9%, respectively; p<0.001). Even when looking at the combined measure of CR and CRi, this difference persisted (66.4% in the aza-ven group and 28.3% in the placebo group; p<0.001).32
After a mean follow-up of 43.2 months, mean overall survival was 14.7 months in the aza-ven group and 9.6 months in the placebo group (hazard ratio 0.85; 95% CI, 0.47-0.72; p<0.001; see also Figure 6). So even in the long term, treatment of patients with azacitidine-venetoclax leads to a longer survival time than treatment with azacitidine alone.33

Figure 7 Mean overall survival of azacitidine + venetoclax (green) and azacitidine + placebo (red), from randomisation to 54-month follow-up.
Source: DiNardo, et al. 2020.32
Major adverse events were nausea (44.5% in the aza-ven group and 36.8% in the placebo group), grade 3 or higher thrombocytopenia (45% in the aza-ven group and 38% in the placebo group), neutropenia (45.9% in the aza-ven group and 28.5% in the placebo group) and febrile neutropenia (42.8% in the aza-ven group and 18.8% in the placebo group). Serious adverse events occurred in 85.1% of patients in the aza-ven group and 77.1% of patients in the placebo group.33
1
Case study I - 83-year-old man with mNPM1
Casus 1.1
An 83-year-old man with a medical history of hypertension, paroxysmal atrial fibrillation and ankylosing spondylitis presents with fatigue symptoms he has had for two weeks.
Laboratory tests show the following results:
- Leukocytes: 21 x 109/L
- 0.8 x 109 neutrophils/L
- 1 x 109 lymphocytes/L
- 19 x 109 blasts/L
- Haemoglobin: 5.3 mmol/L
- Platelets: 11 x 109/L
Bone marrow examination showed hypercellularity with 90% blasts. Genetic testing showed a loss of the Y chromosome and an NPM1 mutation. Other mutations, particularly in FLT3, NRAS, KRAS and IDH1, were negative.
Which therapy is this patient eligible for? Explain your answer.
Bekijk uw antwoord

Because of his age and medical history, the patient is not eligible for intensive chemotherapy. He could be treated with a hypomethylating agent with the addition of venetoclax. Monotherapy with decitabine or azacitidine is also possible, depending on his wishes and performance. However, the presence of an NPM1 mutation (without FLT3/NRAS/KRAS mutation) makes this leukaemia particularly sensitive to combination therapy with venetoclax.
Casus 1.2
Patients receive a combination therapy of azacitidine and venetoclax on a 28-day cycle. The dosage of the drugs is as follows:
- Azacitidine: 75 mg/m2 from day 1 to day 7
- Venetoclax: 400 mg/day, on a build-up schedule (day 1: 100 mg, day 2: 200 mg, day 3 onwards: 400 mg)
On day 21 of the first cycle, bone marrow is collected to assess response and possible bone marrow toxicity. Less than 5% blasts are found in an otherwise hypoplastic bone marrow, after which venetoclax is discontinued.
Can you start a second cycle with azacitidine and venetoclax, and, if so, when?
Bekijk uw antwoord
Yes you can, provided there is recovery of the blood count. The next course of treatment should therefore be postponed. Given there is now remission, you can give GCSF to promote neutrophil recovery.
Casus 1.3
Do you change anything about the frequency of administration and/or dosage of the drugs, and, if so, what?
Bekijk uw antwoord
If the blood count recovers (ANC >1, platelets >75) within six weeks of starting the course, you can give venetoclax again for 21 days with GCSF (after having discontinued venetoclax). The dosage of venetoclax is adjusted only when there are comedications that may increase the level of venetoclax. In particular, consider CYP3A4 inhibitors such as azoles and PgP inhibitors such as ciprofloxacin. Do not change the frequency and dosage of azacitidine.
Casus 1.4
The patient went through the next eight cycles without any problems and showed good blood count recovery. A bone marrow test after the fourth cycle shows complete remission. After the ninth cycle, he developed thrombopenia (grade 3).
What do you do now?
Bekijk uw antwoord
You perform a bone marrow biopsy to rule out a recurrence. This does not appear to be the case here, but you do see signs of increased bone marrow toxicity. You may then consider reducing the duration of venetoclax from 21 to 14 days. Alternatively, if this is insufficient, shortening the azacitidine dosage to 5 days or reducing the dosage can be considered.
Casus 1.5
The treatment schedule now remains unchanged for the remaining cycles. No more side effects occur. After the twelfth cycle there is complete remission, both morphologically and molecularly. The patient is satisfied with their quality of life.
Azacitidine + ivosidenib (in mIDH1)
The efficacy and safety of azacitidine in combination with ivosidenib was investigated in the AGILE study (phase III). Patients with mIDH1 AML who were not fit for intensive therapy were randomised across two groups: azacitidine-ivosidenib (aza-ivo, n=72) and azacitidine-placebo (placebo group, n=74). Both groups were treated with azacitidine (75 mg/m2 SC/IV on days 1-7, in a 28-day cycle). Patients in the aza-ivo group additionally received 500 mg ivosidenib daily, orally; patients in the placebo group received similar placebo treatment. The primary endpoint of the study was disease-free survival, defined as the time from randomisation to treatment failure (failure to achieve remission within 24 weeks), occurrence of relapsed AML, or death.34
At a mean follow-up of 12.4 months, disease-free survival was significantly higher in the aza-ivo group than in the placebo group (hazard ratio 0.33; 95% CI, 0.16-0.69; p=0.002; see also Figure 8a). The estimated probability that a patient would remain disease-free in the first year of treatment was 37% in the aza-ivo group and 12% in the placebo group. After 24 months, mean overall survival was higher in the aza-ivo group (24.0 months) than in the placebo group (7.9 months) (hazard ratio 0.44; 95% CI, 0.27-0.73; p=0.001; see also Figure 8b).34
After a mean follow-up of 28.6 months, mean overall survival was 29.3 months in the aza-ivo group and 7.9 months in the placebo group (hazard ratio 0.42; 95% CI, 0.27-0.65; p<0.0001). In the aza-ivo group, more patients made the transition from 'transfusion-dependent' to 'no longer transfusion-dependent' than in the control group (53.8% versus 17.1%, respectively; p=0.0004).35

Figure 8a (left) Mean disease-free survival of azacitidine + ivosidenib (green) and azacitidine + placebo (red), from randomisation to 30-month follow-up.
Figure 8b (right) Mean overall survival of azacitidine + ivosidenib (green) and azacitidine + placebo (red), from randomisation to 36-month follow-up.
Source: Montesinos, et al. 2022.34
Common adverse events grade 3 or higher were febrile neutropenia (28% in the aza-ivo group and 34% in the placebo group) and neutropenia (27% in the aza-ivo group and 16% in the placebo group). Bleeding was more common in the aza-ivo group than in the placebo group (41% and 29%, respectively). The rate of differentiation syndrome was also somewhat higher in the ivosidenib group (14% vs 8%). By contrast, infections were more common in the placebo group (49%) than in the aza-ivo group (28%).34 The AGILE study has been criticised for switching primary endpoints during the study and for discontinuing the study due to a positive effect, and thus a perceived unethical treatment for the placebo group, after an unspecified interim analysis.36
2
Case study II - 72-year-old woman with AML
Casus 2.1
A 72-year-old woman reports with symptoms of long-standing fatigue, which has become progressive in recent months. In addition, she is known to have chronic cough symptoms and has had a cold for weeks now. Her medical history is as follows:
2019: right total hip
2021: cT2bN0M0 renal cell carcinoma (r), requiring nephrectomy
Moderate COPD with FEV1 60%
In the case history, the patient indicated that she could still perform ADL independently. She does household chores with her husband. She was still able to exercise for an hour and a half a week until recently, but has been unable to do so for two months.
On exploratory labs, the GP found an abnormal blood count with blasts in the differentiation.
- Leukocytes: 10.9 x 109/L
- Blasts: 7.88
- Segments: 0.78
- Haemoglobin: 6.1 g/dL
- Platelets: 68 x 109/L
- Creatinine: 117 µmol/L
What diagnostics do you deploy now?
Bekijk uw antwoord
You perform immunophenotyping studies to distinguish between AML and ALL. In addition, this can collect information on the presence of suitable LAIPs to use as MRD marker in any follow-up test. Note: this requires a more extensive immunophenotyping panel than the usual panel to make the diagnosis.
You also perform cytogenetic tests and molecular tests to detect any mutations. If you know in advance that the patient is not eligible for potentially curative therapy (intensive chemotherapy or allogeneic stem cell transplantation), the deployment of comprehensive genetic diagnostics has only prognostic value, with the exception of the IDH1 determination, which has therapeutic implications in patients who are not suitable for intensive therapy. As there are sufficient blasts in peripheral blood, you can use this test on both blood and bone marrow.
Casus 2.2
The diagnostic tests yield the following results:
Immunophenotyping: blasts of myeloid origin with the presence of aberrant expression of CD7 consistent with acute myeloid leukaemia.
Cytogenetic testing: 48,XX,+X,+8 [10].
Molecular research: IDH1-R132H (VAF 25%), ASXL1 (VAF 31%), RUNX1 (VAF 31%), SRSF2 (39%)
What diagnosis can you make based on this data, and what risk profile is associated with it?
Bekijk uw antwoord
AML myelodysplasia-related (WHO) or AML with myelodysplasia-related gene mutation (ICC) with adverse risk profile (ELN 2022).
Casus 2.3
Is the patient eligible for intensive therapy? Why yes/no?
Bekijk uw antwoord
No. Given her advanced age and present comorbidities (age, previous malignancy and FEV1 <65% with HCT-CI 7), there is a higher risk of early therapy-related mortality (TRM) with intensive chemotherapy and/or allogeneic stem cell transplantation.
Casus 2.4
Given her age and comorbidities (FEV1 <65% with HCT-CI 7), the patient is not eligible for intensive therapy.
What treatment are you putting in place?
Bekijk uw antwoord
Given the presence of an IDH1 mutation, start azacitidine-ivosidenib. In the new AML guideline, this combination has become preferred to azacitidine-venetoclax in mIDH1 AML.
Casus 2.5
Start azacitidine-ivosidenib. Due to nausea during treatment, patient is also treated with ondansetron and metoclopramide.
What are you paying attention to?
Bekijk uw antwoord
Ivosidenib is a potential QTc-prolonging agent, especially in combination with other QTc-prolonging agents such as ondansetron and ciprofloxacin. Metoclopramide plays no role in this context. You will have an ECG performed weekly with each treatment with ivosidenib in the first three weeks of treatment, and if the QTc <480 ms stays the same, it will be done monthly. You will see a normal QTc time, so treatment with azacitidine-ivosidenib can be continued.
Casus 2.6
After two weeks, the patient presented to the ER due to fever and dyspnoea. She has a non-productive cough. The temperature measured in the ER is 39.1°C. Her saturation is 89% on room air with a respiratory rate of 16/bpm. Labs revealed anaemia (Hb 6.2 mmol/L), platelets 45 x 109/L and leukocytes 12.4 x 109/L with 0.9 blasts and increased neutrophils 6 x 109/L and monocytes 2.7 x 109/L. Furthermore, there is a slight exacerbation of her renal insufficiency (creatinine 130 umol/L) and elevated LDH 670 U/L. Thoracic radiograph shows infiltrative abnormalities on both sides.
What is your differential diagnosis?
Bekijk uw antwoord
The clinical picture and timing of symptoms could fit well with differentiation syndrome. This is a serious and potentially life-threatening side effect of IDH inhibitors and should be at the top of your differential diagnosis. Of course, there may also be pneumonia or decompensatio cordis.
Casus 2.7
Bekijk uw antwoord
Start corticosteroids, e.g. dexamethasone 10mg 2dd. If the leukocyte count exceeds 25 x 109/L or this has increased by >15 from baseline, then there is an indication to start hydroxycarbamide, e.g. 2-3 gr divided into 2-3 daily doses. Other supportive measures such as furosemide, dialysis and intubation depend on the clinical picture. If symptoms persist despite adequate therapy, then the IDH inhibitor should be interrupted. If patients improve after administering corticosteroids, these should be tapered off only after significant clinical improvement has occurred and there is a stable situation.37-39
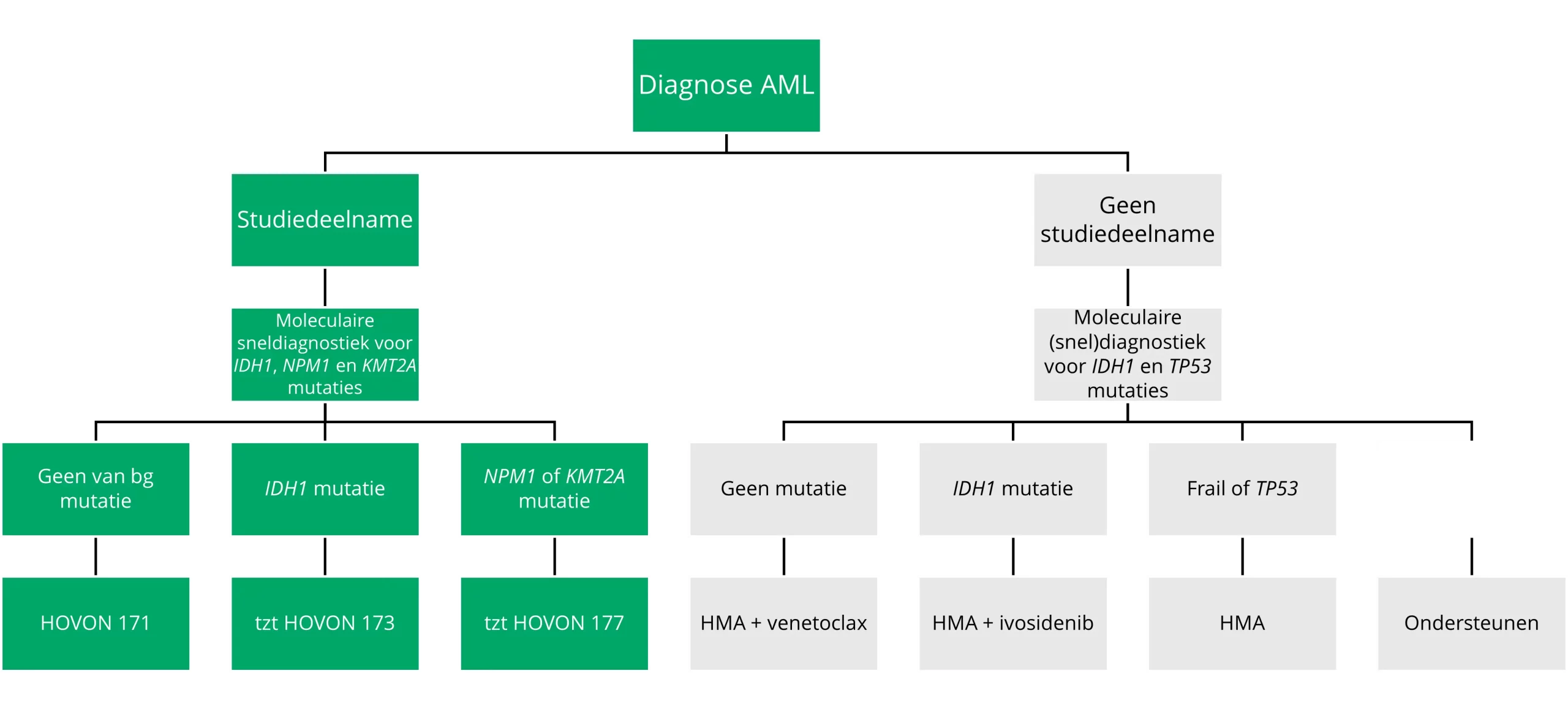
Figure 9 9 Decision tree treatment for non-fit patients (varies by country).
Treatment for refractory disease or recurrent AML
When a patient does not achieve remission after first-line treatment, a switch can be made to second-line treatment. There are different treatment options for fit patients and for non-fit patients. All options are shown in Table 8.
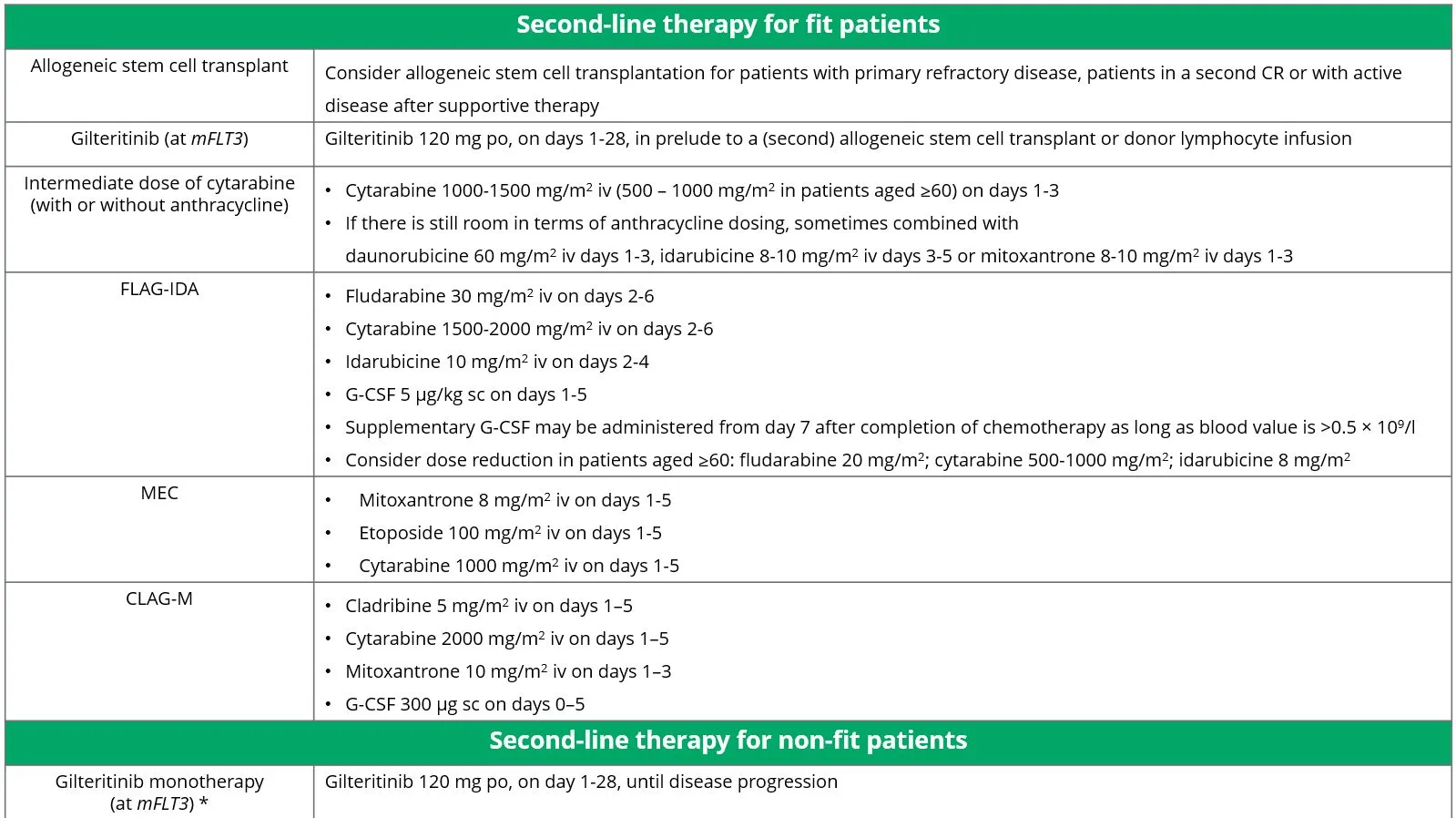
Table 8 Treatment options for patients with refractory and relapsed AML.
Source: Döhner, et al. 2022.20
Currently, there is specific second-line treatment only for patients with mFLT3 AML. This treatment is discussed in more detail below. Studies on other specific treatments for patients with refractory or relapsed AML are ongoing (see also the section ‘The future and new developments')
Gilteritinib monotherapy (in mFLT3)
The efficacy and safety of gilteritinib monotherapy in relapsed or refractory AML with mFLT3 was investigated in the ADMIRAL study (phase III). Patients were randomised across two groups. Patients in the gilteritinib group (n=247) received 120 mg gilteritinib (orally) daily. Patients in the control group (n=124) received supportive chemotherapy. The primary endpoints of the study were overall survival and the percentage of patients with CR or CRi. Secondary endpoints were disease-free survival and the percentage of patients with CR.40
Mean overall survival after 17.8 months was significantly higher in the gilteritinib group than in the control group (9.3 months versus 5.6 months, respectively: hazard ratio 0.64; 95% CI, 0.49-0.83; p<0.001; see also Figure 10). Median disease-free survival was 2.8 months in the gilteritinib group and 0.7 months in the control group. In the gilteritinib group, 34% of patients achieved CR or CRi. In the control group it was 15.3%. When only complete remission (CR) was considered, the rates were 21.1% and 10.5%, respectively.40 After a mean follow-up of 37.1 months, the difference in mean overall survival between the two groups remained apparent. The estimated 2-year survival was 20.6% in the gilteritinib group and 14.2% in the control group.41 Patients with long-term survival in both the gilteritinib and control groups mostly underwent allogeneic stem cell transplantation followed by maintenance therapy with gilteritinib.
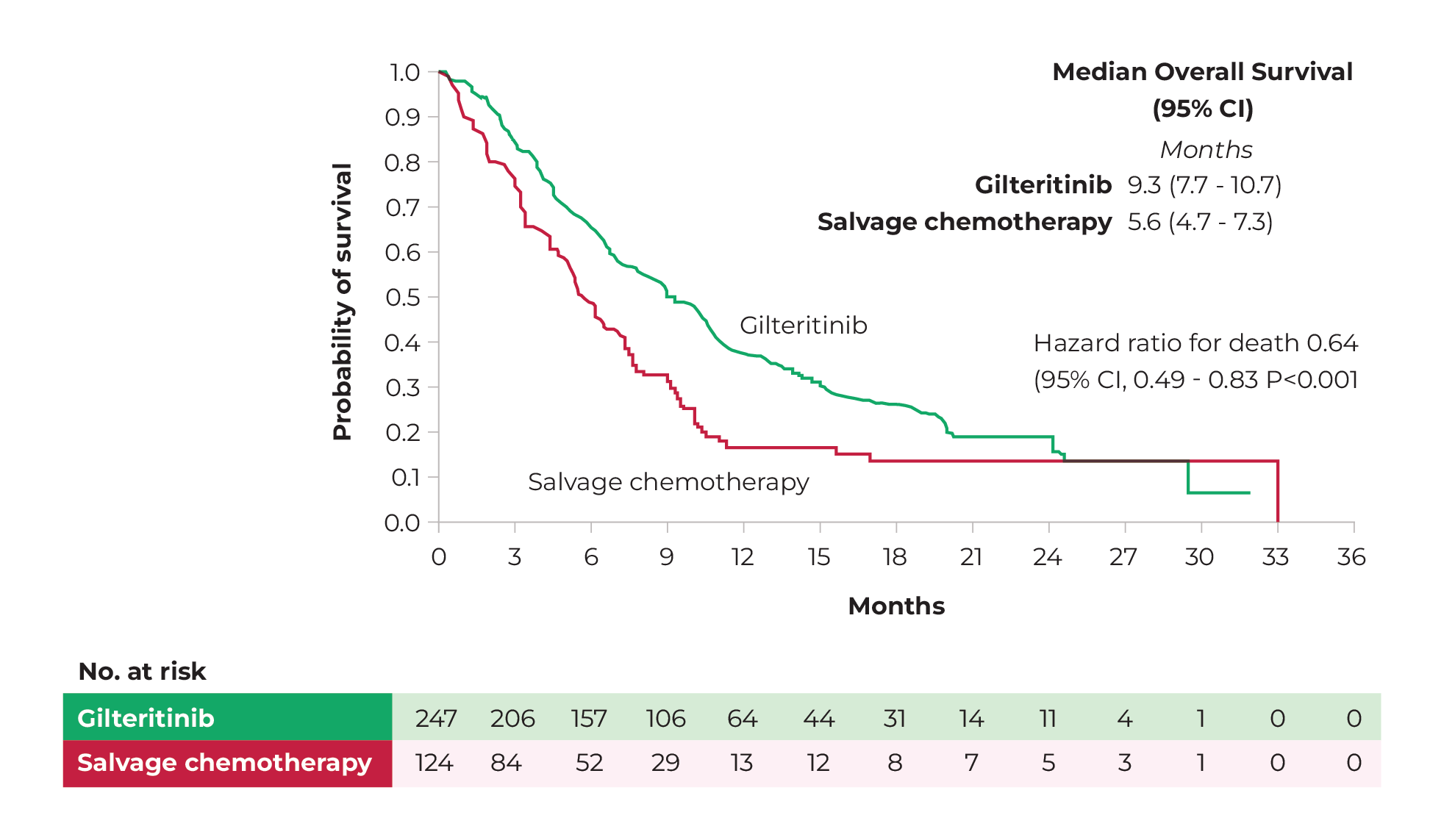
Figure 10 Mean overall survival of gilteritinib monotherapy (green) and supportive chemotherapy (red), from randomisation to 36-month follow-up.
Source: Perl, et al. 2019.40
Adverse events grade 3 and above and serious adverse events were less frequent in the gilteritinib group than in the control group. The most common side effects grade 3 and higher in the gilteritinib group were febrile neutropenia (45.9%), anaemia (40.7%) and thrombocytopenia (22.8%).40 Long-term results also showed that therapy with gilteritinib, especially in the first year, leads to increased transanimase levels in the liver.41
3
Case III - 56-year-old woman with mFLT3-ITD42
Casus 3.1
A 56-year-old woman with no medical history is admitted with fever and general weakness. Physical examination reveals no abnormalities. A complete blood test shows the following picture:
- Leukocytes: 50.5 x 109/L
- Haemoglobin: 8.7 g/dL
- Platelets: 124 x 109/L
Blasts are present in a peripheral blood smear. Further bone marrow examination shows hypercellularity with 80% blasts with myelomonocytic maturation. The results of immunophenotyping and molecular diagnostics are shown in the table below.

What is your diagnosis and risk rating?
Bekijk uw antwoord
The diagnosis is acute myelomonocytic leukaemia (WHO) or AML not otherwise specified (ICC) with an intermediate risk profile (ELN 2022).
Casus 3.2
What treatment are you putting in place?
Bekijk uw antwoord
Leukocytosis is treated immediately with a combination of hydroxycarbamide, allopurinol and IV hyperhydration, pending genetic analysis. After finding out about the mFLT3, a standard induction 7+3 chemotherapy regimen of daunorubicin and cytarabine plus midostaurin was initiated.
Casus 3.3
Complete remission was achieved after the first induction course. Patient then underwent a second course of treatment followed by an allogeneic stem cell transplant with an HLA-compatible sibling. This course was uncomplicated, and the patient experienced no symptoms of graft-versus-host disease. Eight months after transplantation, unfortunately, recurrent AML with rapidly increasing blasts in peripheral blood occurred.
What do you do now?
Bekijk uw antwoord
You perform full cytogenetic and molecular tests again. The same FLT3-ITD is found without additional mutations or cytogenetic abnormalities.
Casus 3.4
What is your treatment plan?
Bekijk uw antwoord
Given relapsed FLT3-positive leukaemia with the possibility of its consolidation with a second allogeneic transplant or DLI, patient is eligible for treatment with gilteritinib.
Casus 3.5
Patient is treated in the second line with gilteritinib at a dose of 120 mg, once a day. Because of the rapidly progressive blastic rise, additionally start hydroxycarbamide. Over the weeks, you can taper off and discontinue hydroxycarbamide. Continue gilteritinib at the standard dose. Two months later, bone marrow evaluation shows complete remission with incomplete blood count recovery. Patient is in good condition, and you decide to give a DLI in order to consolidate the good response.
The future and new developments
Research into new therapies and new combinations of existing drugs remains essential to improve survival of AML patients. This section discusses a number of developments that may play a role in the treatment of AML in the relatively near future.
Fully oral therapy
Decitabine-cedazuridine
Oral decitabine-cedazuridine (ASTX727) is an alternative treatment option for patients who are not fit for intensive chemotherapy. ASTX727 is pharmacodynamically similar to intravenous decitabine. One major difference, however, is that ASXT727 can be given fully orally. The efficacy and safety of ASXT727 compared to decitabine IV in patients with AML was investigated in the ASCERTAIN study (phase III). 87 patients, not fit for intensive chemotherapy, were randomised and treated in two different groups. The mean age of the patients was 78 (61-92 years). 33 patients had an adverse risk classification, 45 patients had an intermediate risk classification, and the risk classification of nine patients could not be determined. Patients in group A received ASXT727 (35 mg decitabine and 100 mg cedazuridine) in cycle 1 of treatment and decitabine IV at a dose of 20 mg/m2 in cycle 2. Patients in group B received decitabine IV in cycle 1 and ASXT727 in cycle 2. All patients were administered ASXT727 from cycle 3 onwards. The primary endpoint of the study was area under the curve (AUC) for equivalence of both treatment options after five days of dosing. The result was an AUC ratio of 99.64% (90% CI; 91.23%-108.8%). After a mean follow-up of 24.5 months, mean overall survival was about 8.9 months (95% CI: 6.0, 13.1). The composite response rate (CR + CRi) was 27.6%, with 19 patients achieving CR and five patients achieving CRi. The safety profile of ASXT727 was similar to that of decitabine IV.43,44
Phase II studies in a small number of patients not fit for chemotherapy (FL, n=42) or with refractory/recurrent AML (RR, n=10) showed that ASXT727 can be combined with venetoclax. Patients received 400 mg venetoclax (PO) every day and 35/100 mg decitabine-cedazuridine (PO) for five days on a 28-day cycle. After a mean follow-up of 12.8 months, overall survival was 12.7 months in the FL group and 7.6 months in the R/R group. Among patients who achieved CR or CRi (26 in the FL group and 5 in the R/R group), recurrence-free survival was 9.8 months in the FL group and 4.6 months in the R/R group. The overall response rate was 67% in the FL group and 50% in the RR group. The most common grade 3/4 adverse events were neutropenic fever (23%), pneumonia (13%), bacteraemia (8%), cellulitis (6%), sepsis (6%) and respiratory failure (6%). The combination of ASXT727 with venetoclax may allow for a full oral therapy for AML in the future.45,46
Oral azacitidine-venetoclax
The efficacy and safety of oral azacitidine in combination with venetoclax is currently being investigated in the VIALE-M study (phase III) as maintenance therapy for patients aged 18 and older. The study evaluates whether the combination of oral azacitidine and venetoclax improves disease-free survival compared with oral azacitidine monotherapy (the current treatment option). The first results are expected in 2025.47
Agents against specific genetic mutations
New FLT3 inhibitors - Quizartinib
The efficacy and safety of quizartinib for the treatment of patients with mFLT3-ITD was investigated in the QuANTUM-First study (phase III). Patients aged 18-75 with mFLT3-ITD AML were randomised across two groups. Patients in both groups were treated with a standard 7+3 induction regimen with cytarabine 100 or 200 mg/m2 per day IV on days 1-7 and anthracycline (daunorubicin 60 mg/m2 per day or idarubicin 12 mg/m2 per day) IV on days 1-3. Patients in the quizartinib group (n=268) additionally received quizartinib 400 mg PO daily, starting on day 8 for two weeks. Patients in the placebo group (n=271) received similar placebo treatment. In addition, quizartinib was used in the quizartinib group as consolidation therapy in combination with high-dose cytarabine and as mono-maintenance therapy. The primary endpoint of the study was overall survival.48
Mean overall survival was 31.9 months in the quizartinib group and 15.1 months in the placebo group (hazard ratio 0.78, 95% CI; 0.62-0.98, p=0.032). A similar proportion of patients in both groups had at least one adverse event (100% in the quizartinib group versus 99% in the placebo group). Adverse events grade 3 or higher were also about equally common in both groups (92% in the quizartinib group and 90% in the placebo group). The most common grade 3 and 4 adverse events were febrile neutropenia, hypokalaemia and pneumonia in both groups, and neutropenia specifically in the quizartinib group.48
Based on these results, quizartinib has now been registered by EMA for the treatment of adult patients with newly diagnosed mFLT3-ITD AML. Due to the slightly higher incidence of bone marrow depression, early mortality and infections, there is a slight preference for the use of midostaurin over quizartinib in the Dutch guideline.1
Menin inhibitors
Several agents are in development that specifically target the protein menin, which plays an important role in gene expression and cell signalling. These inhibitors may be of therapeutic interest for AML patients with mKMT2A and mNPM1. Two of these inhibitors were discussed at the ASH (December 2023): revumenib (SNDX-5613) and JNJ-75276617.
Revumenib
The efficacy and safety of revumenib was investigated in the AUGMENT-101 study (phase I). 68 patients were randomised across two arms. In both arms, patients received revumenib (orally, with or without a strong CYP3A4 inhibitor) every 12 hours on a 28-day cycle. The study included 46 patients with mKMT2A and 14 patients with mNPM1. The overall response rates in these groups were 59% and 36%, respectively. Overall, 33% of patients with mKMT2A and 21% of patients with mNPM1 achieved a CR/CRh or CRi. Therapy-related adverse events occurred in 78% of treated patients. The most common side effects were prolongation of QT interval (56%), nausea (50%), vomiting (40%) and febrile neutropenia (31%). The main adverse events grade 3 or higher were febrile neutropenia (31%), thrombocytopenia (19%) and sepsis (18%).49
JNJ-75276617
The efficacy and safety of JNJ-75276617 was investigated in a phase I clinical trial. The study included 58 patients with refractory or relapsed AML, 33 with mKMT2A AML and 25 with mNPM1 AML. Participants were treated with JNJ-75276617 monotherapy, orally, on a 28-day cycle. Different dosing regimens (once or twice daily) were included in thestudy.50
Treatment could be evaluated in 41 of the patients. In 26 participants, there was evidence of decreased disease in the bone marrow. In 16 patients, this involved a drop of ≥50% in the number of blasts in the bone marrow (PR or better). In patients with the highest dose of JNJ-75276617 (90 mg, n=8), the overall response rate was 50% (two mKMT2A and two mNPM1). One patient achieved CR, one patient CRh and two patients CRi. In patients with a lower dose of JNJ-75276617 (≥45 mg, n=20), the overall response rate was 40% (five mNPM1 and three mKMT2A). Three patients achieved CR, one patient CRh and three patients CRi. The average time to first response in this group was 1.8 months.50
Further examination of biological activity among response patients (n=12) showed that, as expected, there was reduction in expression of menin-KM2TA genes (such as MEIS1 HOXA9 and FTL3) and induction of genes associated with differentiation (ITGAM and MNDA).
52% of treated patients had at least one therapy-related adverse event. The most common was differentiation syndrome (n=8, 14%). Adverse events grade 3 and above occurred in 29% of patients. Neutropenia (n=6, 10%), anaemia (n=4, 7%) and thrombocytopenia (n=4, 7%) were the most common. Research into the optimal dose of JNJ-75276617 for larger trials is ongoing.50
Triple therapy
Currently, most patients who are not fit for intensive chemotherapy get treated with a combination therapy: azacitidine-venetoclax or azacitidine-ivosidenib. Triple treatment options may be added in the future. The following combinations are currently under investigation:
Azacitidine + ivosidenib + venetoclax (aza-ivo-ven)
The safety and efficacy of aza-ivo-ven was investigated in a phase Ib/II study in patients with mIDH1 AML (n=31). Composite CR was 90% with this combination, compared with 83% with ivo-ven. Of the 16 patients in whom it was possible to determine MRD, 63% achieved MRD-negative remission. The IDH1 mutation disappeared in 64% of patients who underwent more than five treatment cycles (n=14). Mean disease-free survival and overall survival were 36 months [94% CI, 23-NR] and 42 months (95% CI, 42-NR), respectively. In particular, patients with mutations in signalling genes seemed to benefit from triple therapy with aza-ivo-ven.51
This combination will soon be further investigated in the HOVON 173 trial in patients with mIDH1 AML, where a placebo-controlled treatment is prospectively compared with azacitidine and ivosidenib with or without venetoclax.
Azacitidine + venetoclax + menin inhibitors
Because of the promising results shown by menin inhibitors in patients with refractory or relapsed mNPM1 or KMT2A-rearranged AML, the question now arises as to whether this is also of added value for patients with newly diagnosed leukaemia. Currently, these patients are treated with azacitidine-venetoclax, and patients with an NPM1 mutation generally have a good response to it. It is possible that this good response rate could be further improved by adding the menin inhibitor revumenib. This will soon be investigated in HOVON 177, a study that prospectively compares placebo-controlled azacitidine, venetoclax with or without revumenib.
Oral decitabine-cedazuridine + venetoclax + menin inhibitors
The SAVE study (phase I/II) investigated the combination of revumenib with venetoclax and oral decitabine-cedazuridine in nine patients with mKMT2A (n=8) or mNPM1 (n=1). Efficacy and safety data were similar to those of previous studies. Based on the initial results, further research is now underway to optimise this combination.52
Conclusion
AML is a complex, heterogeneous disease with an extensive range of treatment options that will only continue to expand in the coming years. As genetic research takes off, more and more genetic mutations are known to play a role in the development of AML. Research into specific agents against clinically relevant mutations is leading to new therapeutic options, for both fit and non-fit patients. As a result, the treatment of AML is becoming increasingly personalised.
Of note here is also the development of MRD, which is important in therapy selection and monitoring in addition to its prognostic value. Knowledge of new treatments and monitoring is essential to continue providing the best possible care to patients with AML.